
 日本語
日本語 English
English三芳香基取代吡啶的合成及其应用
Today:0views / Total:2,892views
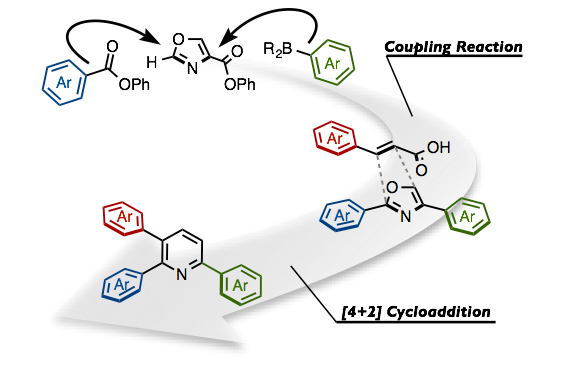
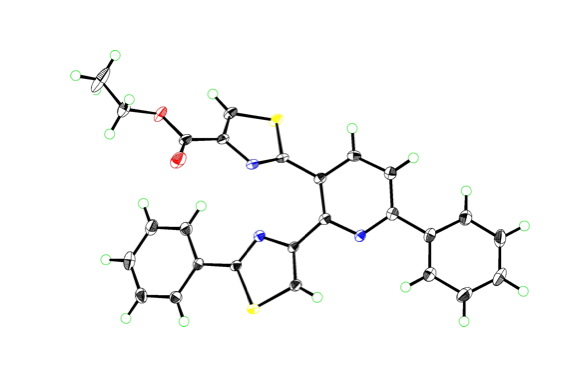
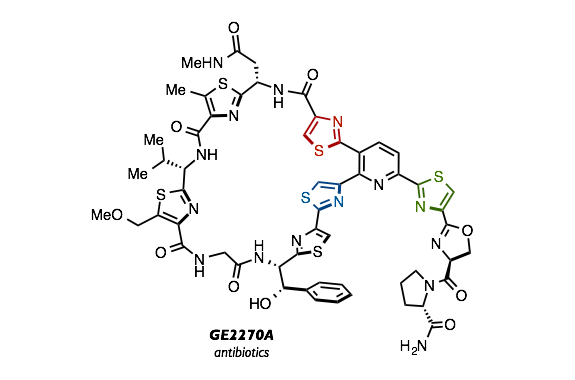
“Synthesis of Triarylpyridines in Thiopeptide Antibiotics by Using a C−H Arylation/Ring-Transformation Strategy”
Amaike, K.; Itami, K.; Yamaguchi, J.
本文研究多取代吡啶骨架构筑的新方法「偶联反应/环化反应」的开发,及用它做关键反应应用在硫肽类抗生素GE2270型、阿米正大克拉霉素类化合物的合成中。
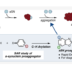
针对4-恶唑羧酸酯类化合物,我们研究室开发的镍催化有机硼酸和芳香酯的偶联反应与第一章讲述的利用镍催化的唑类羧酸酯先脱酯化、再与C–H键芳基化反应联用,合成出2,4-二芳基恶唑。之后,将所得的2,4-二芳基恶唑的与烯丙基丙烯酸作用,发生[4+2]环加成反应、并脱羧、脱氢,位置选择性的得到了2,3,6-三芳基吡啶骨架。通过一系列二芳基恶唑和烯丙基丙酸衍生物相互组合反应,快速合成出15种以上的2,3,6-三芳基吡啶衍生物。
之后我们将开发所得的2,3,6-三芳基吡啶衍生物,应用于硫肽类抗生素的合成中。先将三芳基吡啶合成所必要的4-噻唑基恶唑与噻唑苯酯、噻唑基丙烯酸分别合成出来。将这些化合物用在开发了的三芳基吡啶化合物合成方法中,成功合成出作为硫肽类抗生素合成中间体的2,3,6-三芳基吡啶。之后这个三芳基吡啶中间体经过几步反应后,成功完成了硫肽类抗生素GE2270型、阿米正大克拉霉素类化合物的全合成。



論文ピックアップ


最近の記事
-

2024.4.24
(日本語) 〜サクラ咲く、新たな風の予感〜 新入生歓迎会2024 -
2024.4.23
(日本語) 福山本解説 A1 -

2024.4.22
(日本語) 初めまして、高橋です -

2024.4.19
(日本語) 山口研の歩き方 -

2024.4.18
(日本語) よしかわです
