
 日本語
日本語 English
English- Home
- 分子的结合
分子的结合
人类最小的功能单元「分子」的结合,即分子构建的产生和发展历史已经有200年了。虽然这一研究也历经百年,但是相比起文学、法学以及科学中的机械等等宏观物体的构建方法,它还是一门相对年轻的学科。
在这一学科的发展史中,迄今为止,已经开发出很多种分子构建的方法了。而谈到有机分子的构建,将分子连接起来方法中,其中最有效方法之一就是碳-碳键的构建反应。
例如2010年获得诺贝尔化学奖的研究—偶联反应就是大家都能用到的有机分子构建方法。我们的目标是拓宽偶联反应的范围,开发能够自由自在构建分子的催化反应。
研究实例(进行中)
开发芳香族羧酸衍生物作为偶联反应试剂的高难度转化反应
芳香羧酸衍生物可以说是市售试剂及合成中间体中「无处不在的结构」。芳香羧酸衍生物价格非常低廉,在多环合成方法中,原料中的芳香羧酸(羧酸酯)在反应前后能够保持稳定。如果能将它们不经过其他活性官能团,而直接将羧酸衍生物转变为碳原子或其它杂原子,不仅能够缩短工艺路线,这还将会成为一种新型可持续性的合成战略。另外,现在的催化偶联反应的偶联试剂(亲电试剂)一般都是芳香族卤化物(离去基团:卤素),最近些年,利用较为廉价的酚类衍生物(离去基团:-OR)作为亲电试剂也成为偶联反应的发展趋势之一。另外,目前有报道过利用羧酸作为亲电试剂的合成方法,不过会用到高价钯催化剂,苛刻的反应条件,或者底物适用范围有限,这些都限制其应用范围。因此,我们通过使用羧酸衍生物(离去基团:-CO2R)作为亲电试剂来开发新型偶联反应,意在能扩大偶联反应概念的范围和可能性。
我们开发了一种新型镍催化剂[Ni/dcype催化剂,dcype = 1,2-bis(dicyclohexyl)phosphinoethane],用于1,3-唑类芳香化合物与偶联试剂芳香羧酸酯(Ar-CO2Ph)的新型脱酯型C-H键芳基化反应。 (下图左式)[2]。芳香酯从形式上看脱去酯基,与1,3-唑类化合物发生偶联反应,在此之前,酯作为离去基团的反应从未报道过。利用这一反应,我们成功实现了复杂天然产物muscoride A的全合成。最近,芳香卤化物的替代物–芳香羧酸酯化合物的Suzuki-Miyaura偶联反应也被成功开发(下图右式)。反应体系利用非常便宜的Ni(OAc)2/P(n-Bu)3催化剂和碳酸钠,实现了多种芳香羧酸酯与有机硼类化合物的反应。通过这一反应,简化缩短了一些偶联反应的步骤。这一脱酯型C-H芳基化反应并与之后发现的镍催化剂联用,成功确立了各类分子变换技术的正交反应。
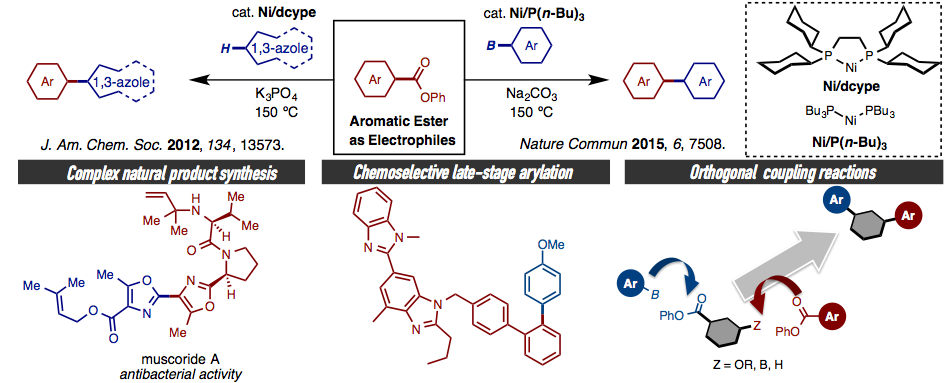
我们之后的研究重点在:将上述两种芳香族酯的偶联反应作为基石,开发并扩大更多潜在的杂环化合物及碳原子骨架作为亲核试剂的反应。也就是说,羧酸衍生物的直接碳碳键构建及碳-杂原子化学键构建的反应。
研究成果
开发直接C–H键偶联反应在生物活性分子中应用的新型合成法
联芳基结构骨架,是医药品及生物活性天然产物最常见的结构骨架。「如何构筑联芳基骨架」也是有机合成化学中最重要的研究课题之一。近年来,C-H键的直接转化芳基化反应作为最理想的联芳基化合物构筑方法,受到业界的广泛关注。我们通过之前的策略,开发新反应・新催化剂,希望能推动这一研究领域的发展。本研究的重点是对生物活性分子的迅速合成非常重要的「真正自由的芳香环连接反应」的开发,及实现这些目标化合物的超短工程合成。研究成果上看,我们合成了10种以上的C-H键直接芳基化的新型偶联反应、15种以上天然产物的生物活性分子的合成以及400种以上的新型杂环联芳化合物衍生物的合成。
以下就几个方面概括介绍一下本研究室在直接的碳-碳键构建反应及C–H键直接转变反应方面的研究成果。
(1) 镍催化的杂芳环C–H键偶联反应
杂芳香环C–H键活化偶联反应是用Pd, Rh, Ru催化剂作用下与芳基化试剂的偶联,通常是卤代芳烃作为主流。本研究中,我们利用便宜的镍催化剂开发了针对3种芳基化试剂 (卤代芳烃、酚类衍生物、芳香族酯)的直接偶联反应。这一镍催化的反应,可以用于痛风治疗药的快速合成、复杂的奎宁化合物的直接C–H偶联反应,在天然产物的收敛合成中也能发挥其巨大作用。另外,我们还成功开发了镍催化的C-H键烯基化、羰基化合物的α-芳基化、有机硼酸和芳香族酯的新型偶联反应。
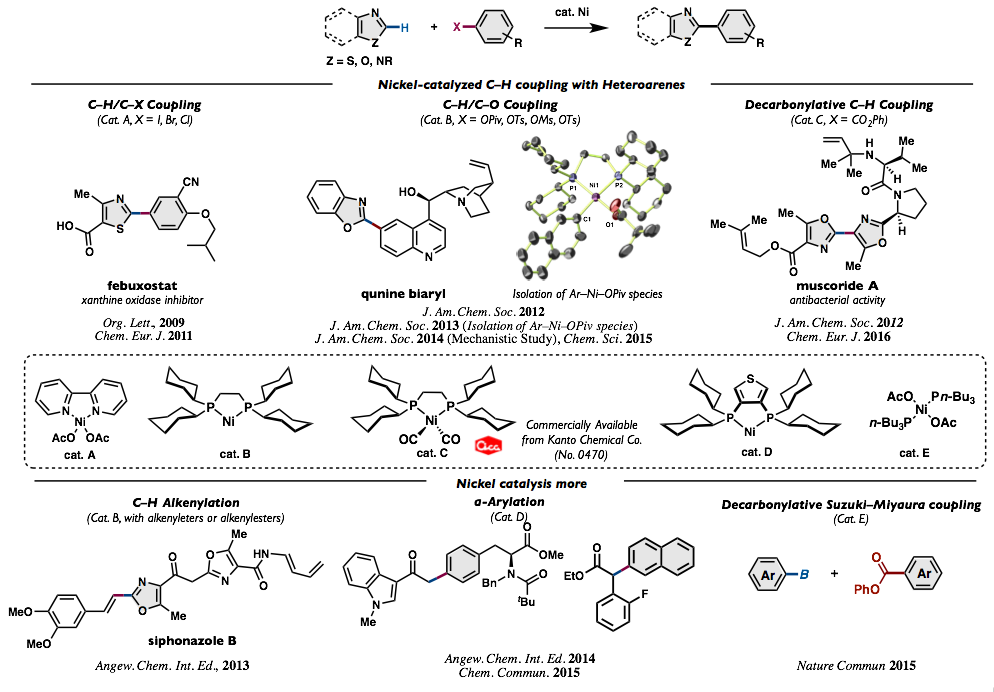
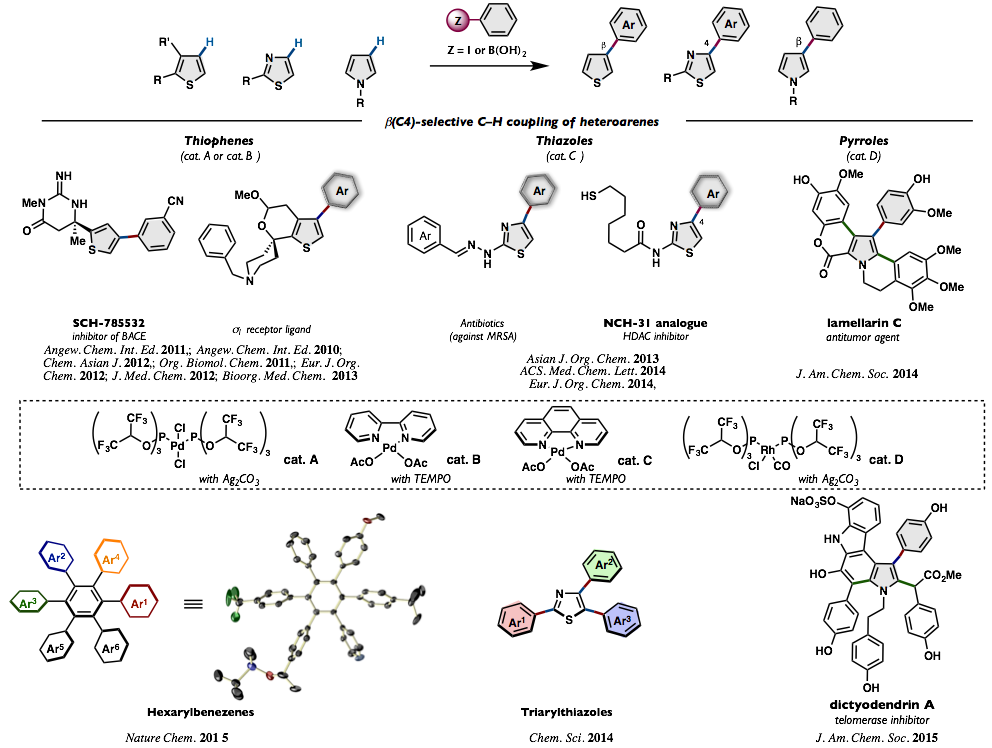
(2) 5元芳香杂环的β位选择性C–H偶联反应
杂芳香环的C-H偶联反应,已经报道了许多例过渡金属催化剂反应。但是C-H键的位置选择性转换,通常依赖于底物的反应性及结构,而通过催化剂来控制位置选择性的例子还从未报道过。基于这一假想,我们开发了能够使噻吩和吡咯的β位选择性芳基化,噻唑的C4位选择性偶联的新型催化剂。开发所得的反应可以用于艾尔兹海默症(老年痴呆症)治疗药及新型σ1受体、MRSA耐性菌的有效化合物;并通过最终阶段C-H键活化偶联得到HDAC抑制剂的衍生物。另外将这类反应作为关键反应,实现了多取代芳基的程序合成、吡咯生物碱的短工程合成。
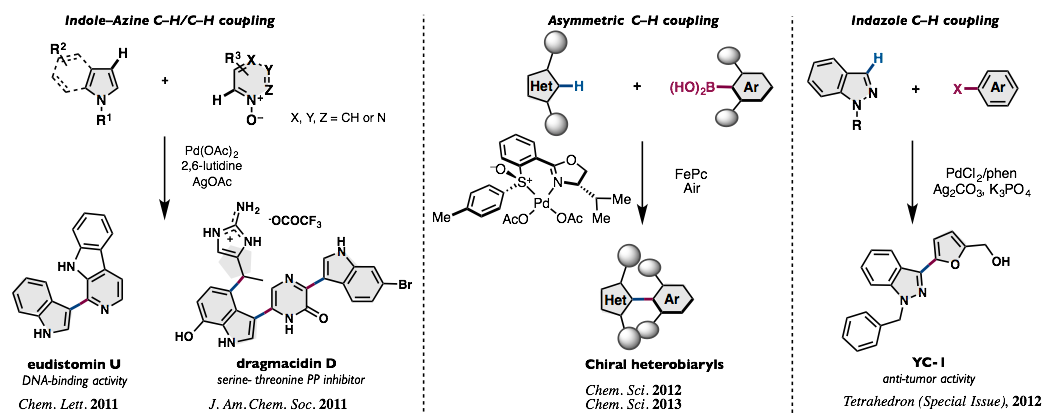
在应用的其他方面,我们还成功实现了
(3)吲哚和嗪类化合物的偶联反应;(4)杂芳香环的不对称C–H键偶联反应;(5)医药化合物重要骨架吲唑的C–H键偶联反应。
以上就是本研究的成果,我们将直接构筑比较困难,而且需要多步反应的碳碳键形成反应,成功实现一步合成。今后,希望这样快捷的基础合成技术能够在天然产物及医药品候补化合物群的高效快速建立及促进其在化学生物学基础研究中应用领域发挥潜在的优势。
